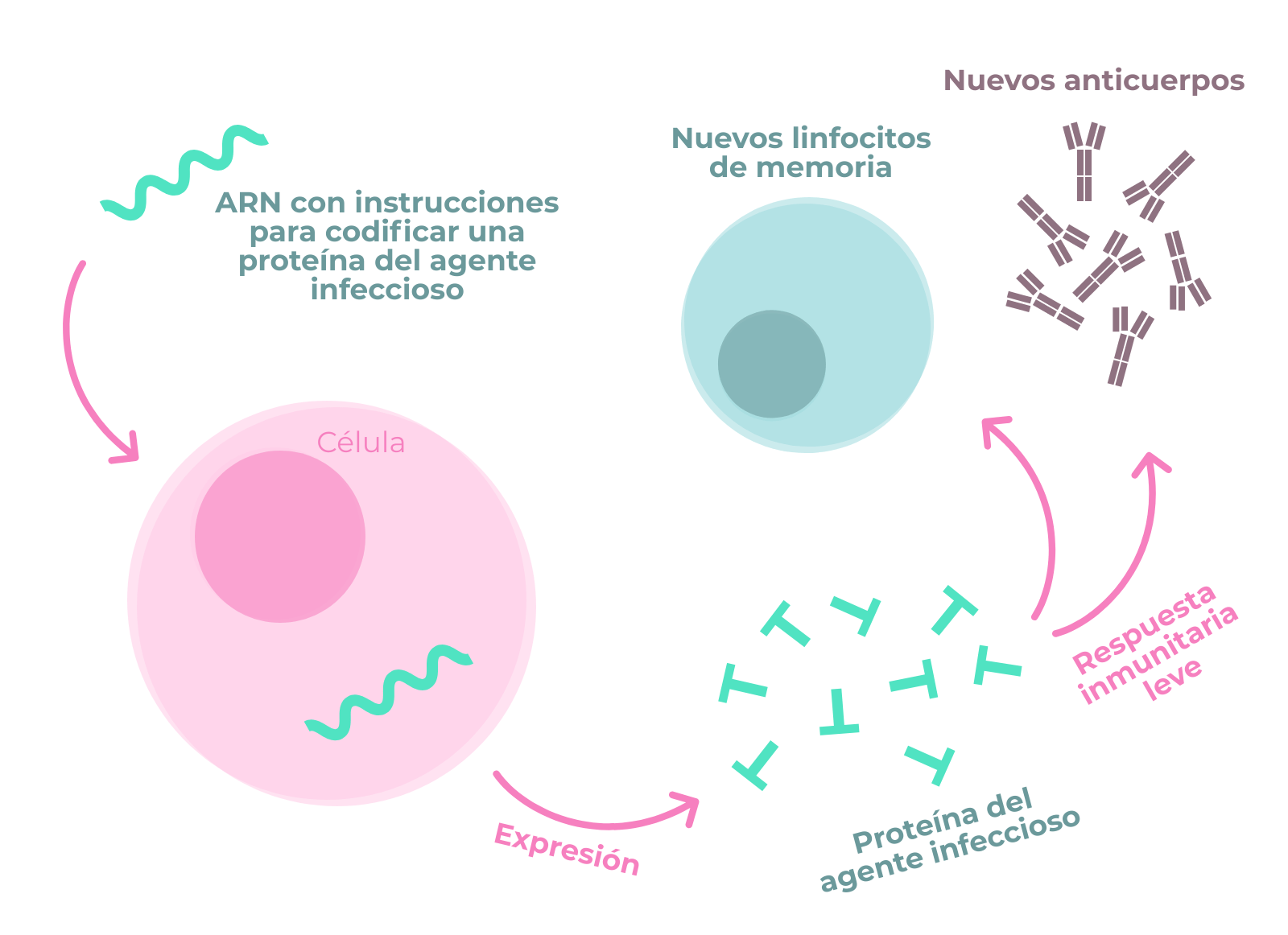
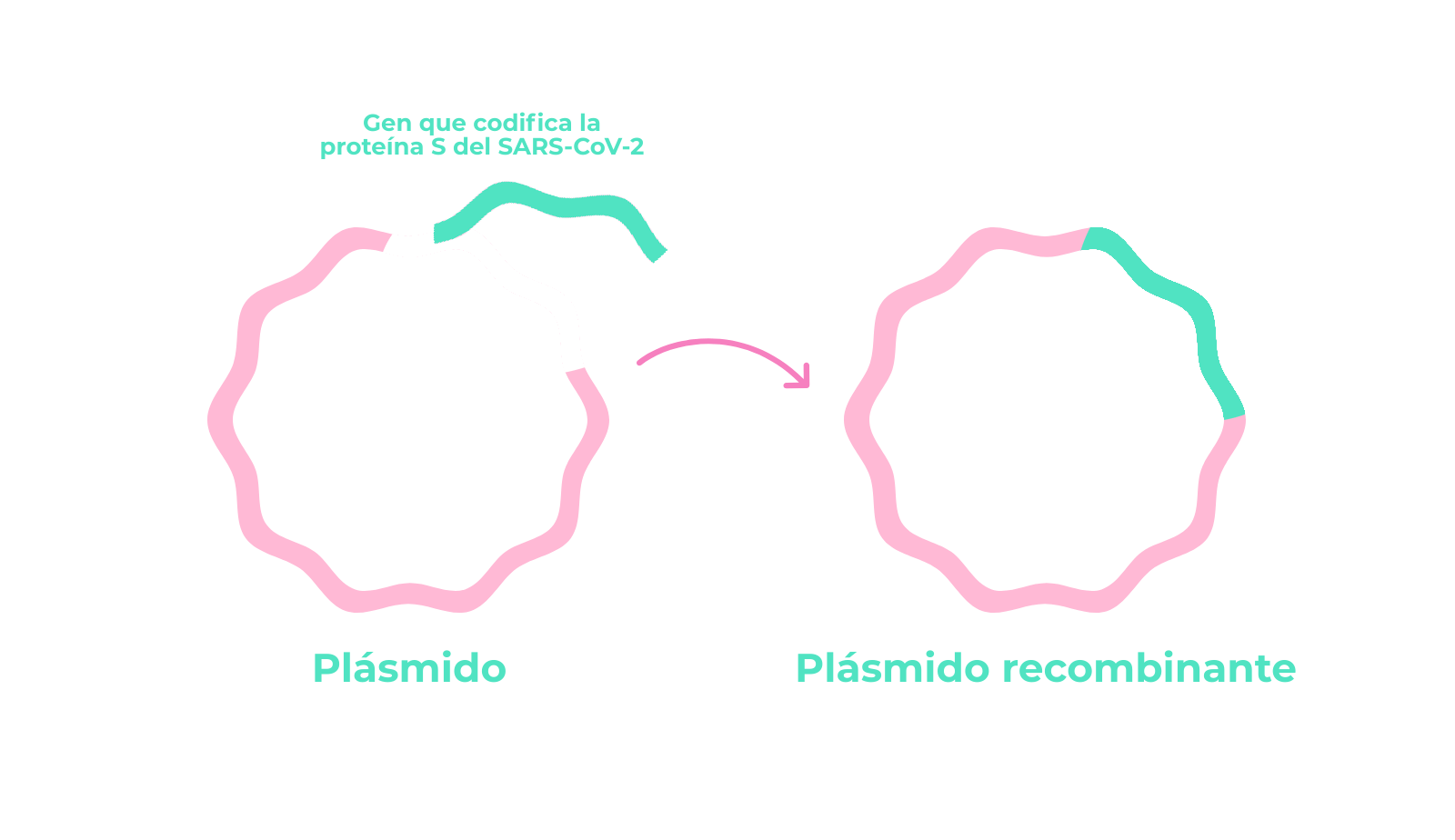
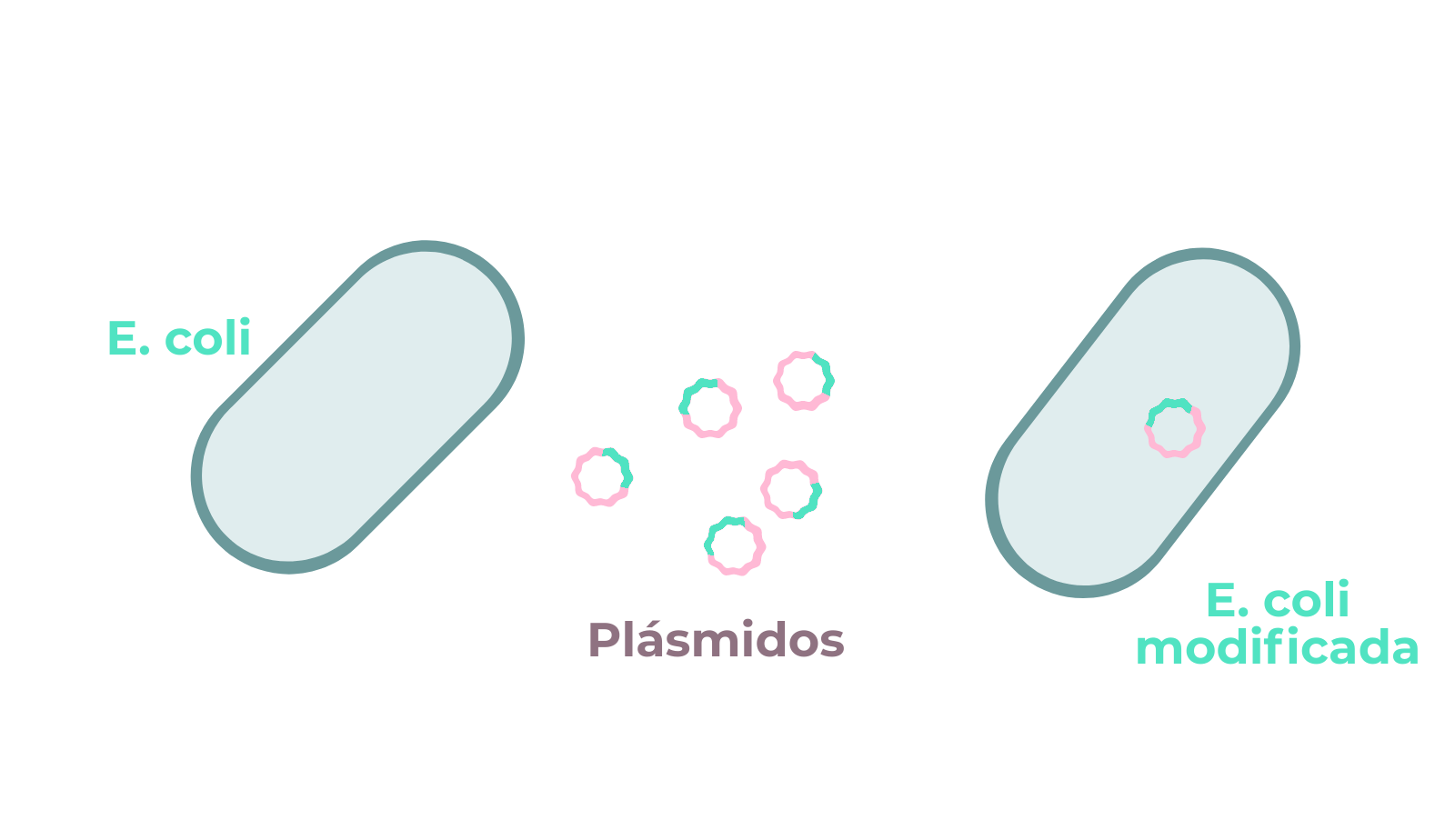
Conoce a tu enemigo. La conocida frase del famoso tratado de estrategia “El Arte de la guerra” adquiere un carácter esencial frente al nuevo agente microscópico que amenaza la salud de miles de personas y pone a prueba la organización y sistemas sanitarios de múltiples países: el coronavirus SARS-CoV-2.
Una de las mejores formas de conocer un organismo, es secuenciar su genoma, que contiene las instrucciones necesarias para hacerlo funcionar. Cuando se produce una pandemia como la de COVID-19, conocer el genoma del agente infeccioso responsable proporciona información con gran relevancia para los investigadores. Les permite identificar qué es lo que causa la enfermedad, conocer su origen y evolución con el tiempo o desarrollar estrategias terapéuticas para hacerle frente.
Identificación de SARS-CoV-2
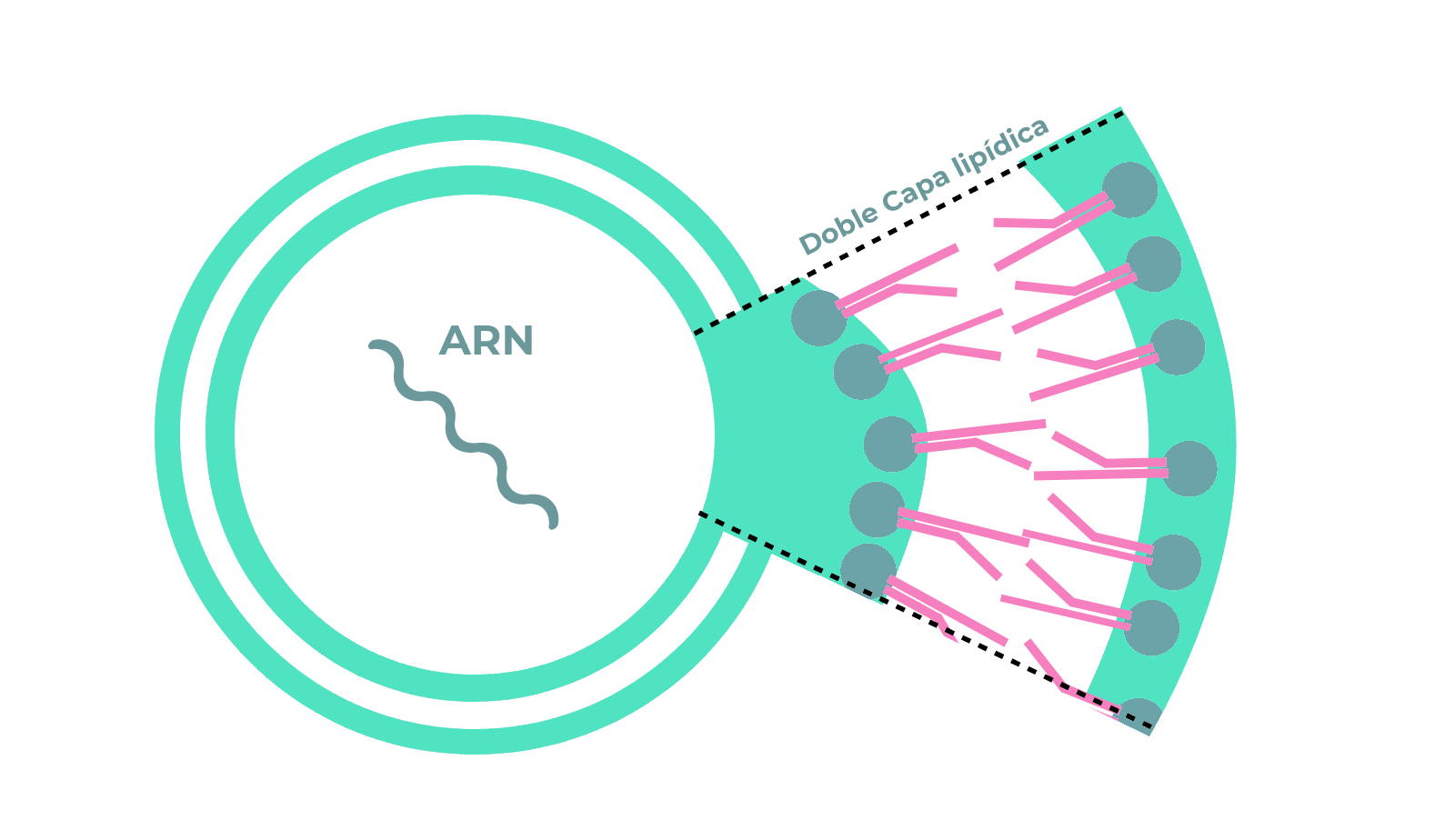
La primera secuencia del genoma del agente infeccioso responsable de la epidemia iniciada en Wuhan se obtuvo en enero. Esta información fue crítica para identificar al virus como un coronavirus, muy similar al coronavirus responsable del Síndrome Agudo Respiratorio Grave (SARS en sus siglas en inglés), enfermedad respiratoria originada en Asia en 2003, que se propagó por diversos países. Los coronavirus son virus de ARN que se reciben su nombre por la característica estructura de su forma infectiva, similar a la corona solar. Este tipo de virus es responsable de muchos de los resfriados comunes, que no tienen consecuencias importantes sobre la salud. No obstante, otros pueden causar enfermedades mortales como el SARS o el síndrome respiratorio de oriente medio (MERS en sus siglas en inglés).
La similitud del nuevo coronavirus con el virus responsable del SARS, denominado SARS-CoV, fue determinante para denominar al nuevo virus SARS-CoV-2.

Características del nuevo coronavirus
El análisis del genoma de SARS-CoV-2, en combinación con las pruebas bioquímicas y las imágenes obtenidas por microscopía electrónica, permite conocer mejor sus características, incluyendo aquellas que pueden ser aprovechadas por los investigadores para desarrollar terapias. Así, a partir de pruebas bioquímicas y estructurales los investigadores han determinado que la parte más variable del genoma del coronavirus se encuentra precisamente en el dominio de unión al receptor de la proteína S, una proteína necesaria para la invasión del virus. En humanos, este dominio proteico tiene una afinidad especial por los receptores ACE2 de las células del hospedador.
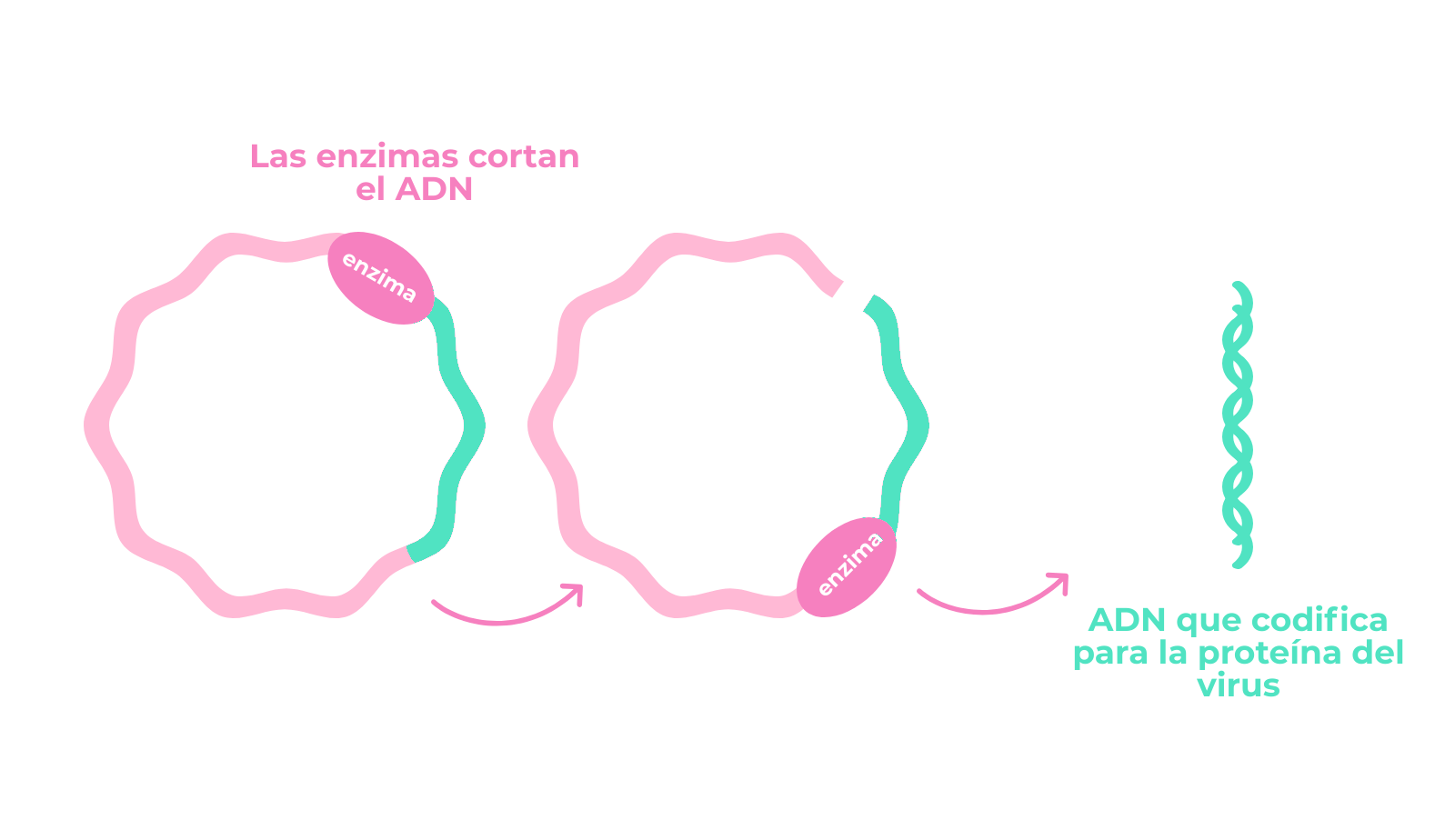
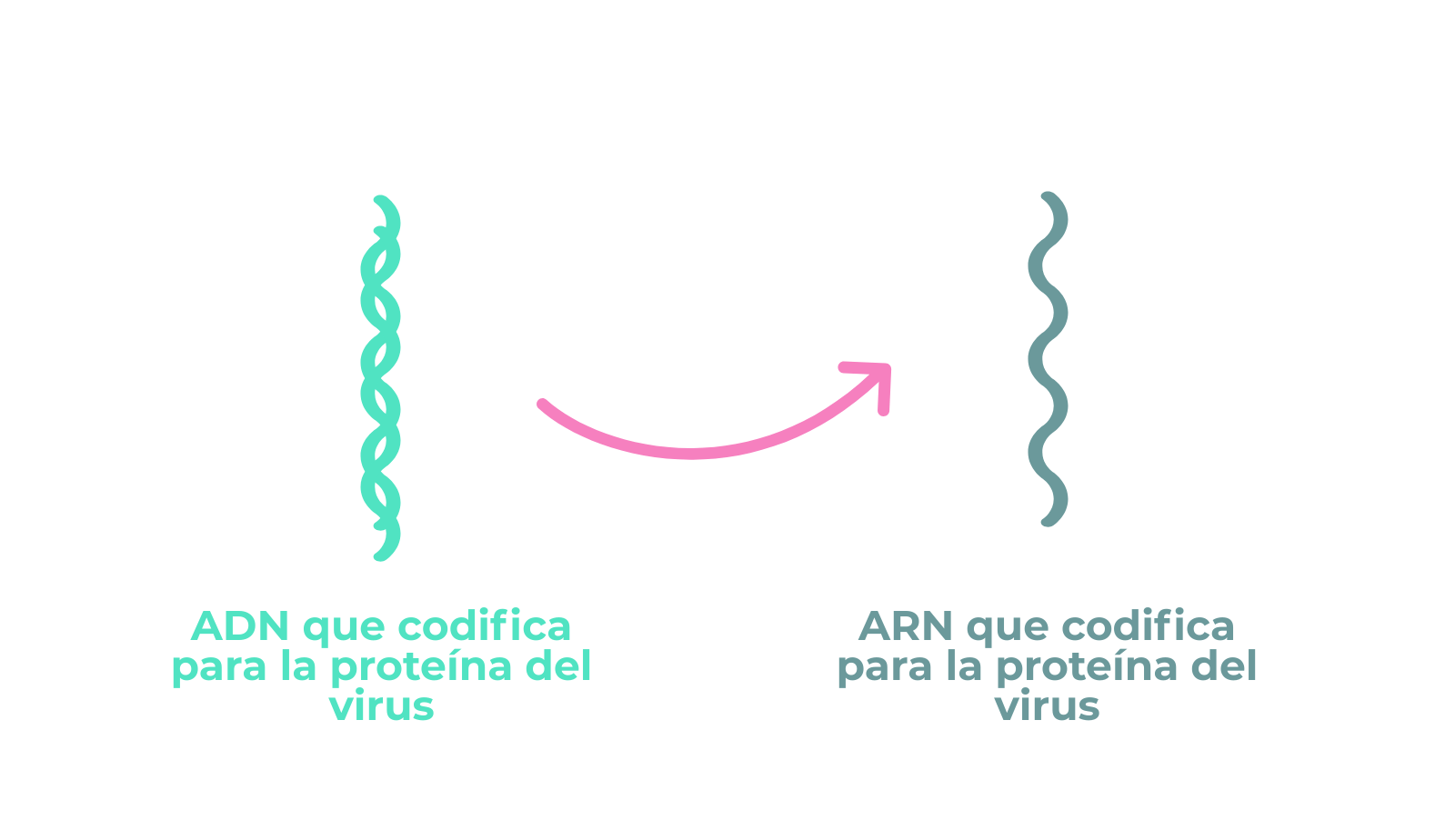
Cuando los coronavirus infectan una célula, liberan en su interior su ARN que puede ser leído por la maquinaria celular para producir una larga cadena polipeptídica que es posteriormente fragmentada en péptidos funcionales para el virus. Otra característica encontrada en el genoma de SARS-CoV-2 es que esa cadena polipeptídica presenta un fragmento en que facilita la separación de los péptidos correspondientes a la proteína S.
Origen del Coronavirus de Wuhan
Una de las cuestiones más discutidas sobre el coronavirus SARS-CoV-2 es su origen. Las primeras investigaciones apuntaban a un posible origen animal, sin que estuviera claro cuál era exactamente. La hipótesis más aceptada en la actualidad es que el virus deriva de un virus de murciélagos que pasó a nuestra especie a través de un intermediario, como ocurrió en el caso de los coronavirus responsables del SARS y del MERS.
En paralelo a los estudios científicos, también empezaron a surgir también diversas teorías sobre si el coronavirus había sido creado en un laboratorio y liberado intencionada o accidentalmente. Un reciente análisis, que considera la información disponible del genoma del virus, descarta la creación del virus y plantea los posibles escenarios de su evolución.
Los autores del análisis indican que es muy poco probable que SARS-CoV-2 haya sido creado a partir de la manipulación de otro coronavirus. La estructura del genoma de SARS-CoV-2 no deriva de la de otro virus, que sería lo que se esperaría si fuera de diseño. Además, aunque el dominio proteico de unión al receptor de la proteína S mencionado anteriormente tiene afinidad por los receptores ACE2 humanos, esta afinidad no está optimizada mediante predicciones. Es diferente de lo que los algoritmos predictivos estimarían.
Los investigadores plantean diversos escenarios posibles de aparición del SARS-CoV-2. En el primero de ellos, el virus habría adquirido sus características a través de la selección natural en una especie animal antes de que el virus saltara a la especie humana. Entre los datos que apoyarían esta posibilidad está el hecho de que la variación del dominio de unión de la proteína S encontrada en SARS-CoV-2 es similar a la observada en otros coronavirus encontrados en pangolines. Sin embargo, los coronavirus más cercanos a SARS-CoV-2 presentes en animales no tienen la secuencia que favorece fragmentación de péptidos de la proteína S.
En el segundo escenario, la selección natural en el virus se habría producido en humanos tras la transferencia del virus desde una especie animal. La adquisición de la secuencia de corte habría sido incorporada también una vez en la especie humana.
Un tercer escenario posible planteado por los investigadores es que los cambios en el genoma de SARS-CoV-2 respecto a otros virus (es decir, su origen) hayan ocurrido por accidente en cultivos celulares de investigación. Los autores del análisis no encuentran evidencias de que ese haya sido el caso e indican que no creen que este escenario sea plausible. No obstante, señalan que con la información actual no se puede concluir de forma definitiva cuál de los tres escenarios es el correcto y será necesario investigar más.

Evolución del coronavirus y seguimiento de la enfermedad
La secuenciación de los genomas de los virus SARS-Cov-2 que se van aislando de las personas diagnosticadas como infectadas permite elaborar un registro de cómo va evolucionando el virus. Una característica de los virus de ARN como SARS-Cov-2 es que mutan rápidamente, y van acumulando cambios en su genoma. Registrar estos cambios es una herramienta epidemiológica muy útil para hacer el seguimiento de la infección.
Desde el inicio de la pandemia de COVID-19 los investigadores están compartiendo las secuencias de SARS-Cov-2 obtenidas en los diferentes países, lo que ha permitido elaborar un mapa dinámico de la evolución de SARS-Cov-2 que permite visualizar la progresión espacial y temporal del virus. Las primeras secuencias de los coronavirus aislados en España fueron incorporadas el pasado viernes, tras ser obtenidas por el Servicio de Secuenciación y Bioinformática de FISABIO y por el grupo de investigación en Epidemiología Molecular del I2SysBio, liderados por Fernando González-Candelas, catedrático de Genética de la Universitat de València.
Con la información proporcionada por los diferentes equipos, se sabe que el virus está mutando y que la tasa de mutación de SARS-Cov-2 es más lenta que la del virus de la gripe. Sin embargo, todavía no hay datos sobre si ha cambiado su virulencia. Conforme se obtengan más secuencias de más pacientes podrá mejorarse la resolución en el mapa de evolución del virus, y obtenerse más detalles.

Diseño de terapias
En la actualidad, el objetivo final de todas las investigaciones sobre el coronavirus SARS-Cov-2 es encontrar una forma de detenerlo y tratar a los pacientes. En este sentido conocer qué regiones del genoma del virus son más o menos susceptibles a tener mutaciones puede ser utilizado en el diseño de estrategias terapéuticas.
Por una parte, la similitud del genoma de SARS-Cov-2 con el del virus SARS-Cov-1 plantea la posibilidad de utilizar o adaptar estrategias que han mostrado potencial en SARS a la enfermedad causada por SARS-Cov-2. Por otra muchas de las aproximaciones para hacer frente a los virus dependen del reconocimiento de moléculas, bien ARN bien proteínas. En este sentido, interesará diseñar los agentes terapéuticos para que reconozcan moléculas con baja probabilidad de sufrir cambios.
En resumen, la secuenciación del genoma de SARS-Cov-2 es una herramienta esencial para estudiar la progresión y evolución del virus, así como para poder desarrollar tratamientos o vacunas para COVID-19.
Referencias:
Andersen KG, et al. The proximal origin of SARS-CoV-2. Nat Med. 2020. https://doi.org/10.1038/s41591-020-0820-9
Liu C, et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent Sci. 2020. Doi: https://doi.org/10.1021/acscentsci.0c00272
Fuentes:
Las primeras secuencias genómicas del virus SARS-CoV-2 de dos pacientes españoles se han obtenido en València. https://www.uv.es/instituto-biologia-integrativa-sistemas-i2sysbio/es/novedades-1285990801509/Novetat.html?id=1286122919661
¿De qué sirve secuenciar el coronavirus? https://theconversation.com/de-que-sirve-secuenciar-el-coronavirus-133717
¿Cómo cambian los virus? https://theconversation.com/como-cambian-los-virus-132996
Fuente: https://genotipia.com/genetica_medica_news/sars-cov/